Research
My goal is to understand and optimize modern “click” reactions using state-of-the-art computational tools. By forming and breaking bonds at molecular level, we can design more efficient, sustainable, and precisely controllable reactions for use in materials science and chemical biology.
My Approach
My work is primarily computational, integrating expertise in quantum chemistry and computer science. By leveraging a suite of powerful tools, we can move beyond trial-and-error and uncover the fundamental principles that govern reactivity. Key techniques I use include:
Density Functional Theory (DFT): To model the electronic structure of molecules and predict their behavior.
Energy Decomposition Analysis (EDA): To break down the forces of a chemical bond into understandable parts (like electrostatic and orbital interactions), revealing why a reaction is favorable.
Ab Initio Molecular Dynamics (AIMD): To simulate the real-time movement and interactions of atoms and molecules during a reaction.
Machine Learning (ML): To analyze complex simulation data and build predictive models that accelerate the discovery of new and improved reactions.
Research Themes
Here are some of the key themes I am currently focused on:

The Nobel Prize-winning concept of “click chemistry” has revolutionized how scientists build molecules. Its hallmark features, modularity, high yields, and broad functional group tolerance, make click reactions invaluable in drug discovery, materials science, and chemical biology. Among the most iconic examples are the Copper-catalyzed Azide–Alkyne Cycloaddition (CuAAC) and Sulfur-Fluorine Exchange (SuFEx) reactions. SuFEx is expanding the synthetic toolbox for chemists seeking to build complexity with precision, so understanding its mechanism and electronic features at the molecular level is crucial and that’s exactly what I aim to do. My work builds on this foundation by exploring and advancing a powerful new variant: the Sulfur(VI)–Phenolate Exchange (SuPhenEx) reaction. By replacing fluoride with a broad range of phenols, SuPhenEx offers a more versatile, synthetically appealing, and environmentally friendly strategy for constructing advanced molecules and materials.
What if click chemistry could be both dynamic and precisely controlled to produce a specific 3D structure (enantiomer)? Our work on the SuPhenEx reaction demonstrated that this is an easy fix. We found that the reaction is not only fast finishing in minutes at room temperature with high yields but is also enantiospecific. This allows for the creation of both enantiomers from a single starting material and can even bypass the need for fluorine chemistry altogether, avoiding issues like racemization. By combining the precision of click chemistry with dynamic covalent behavior, SuPhenEx opens new doors for asymmetric synthesis and the design of stable, yet degradable, polymers.
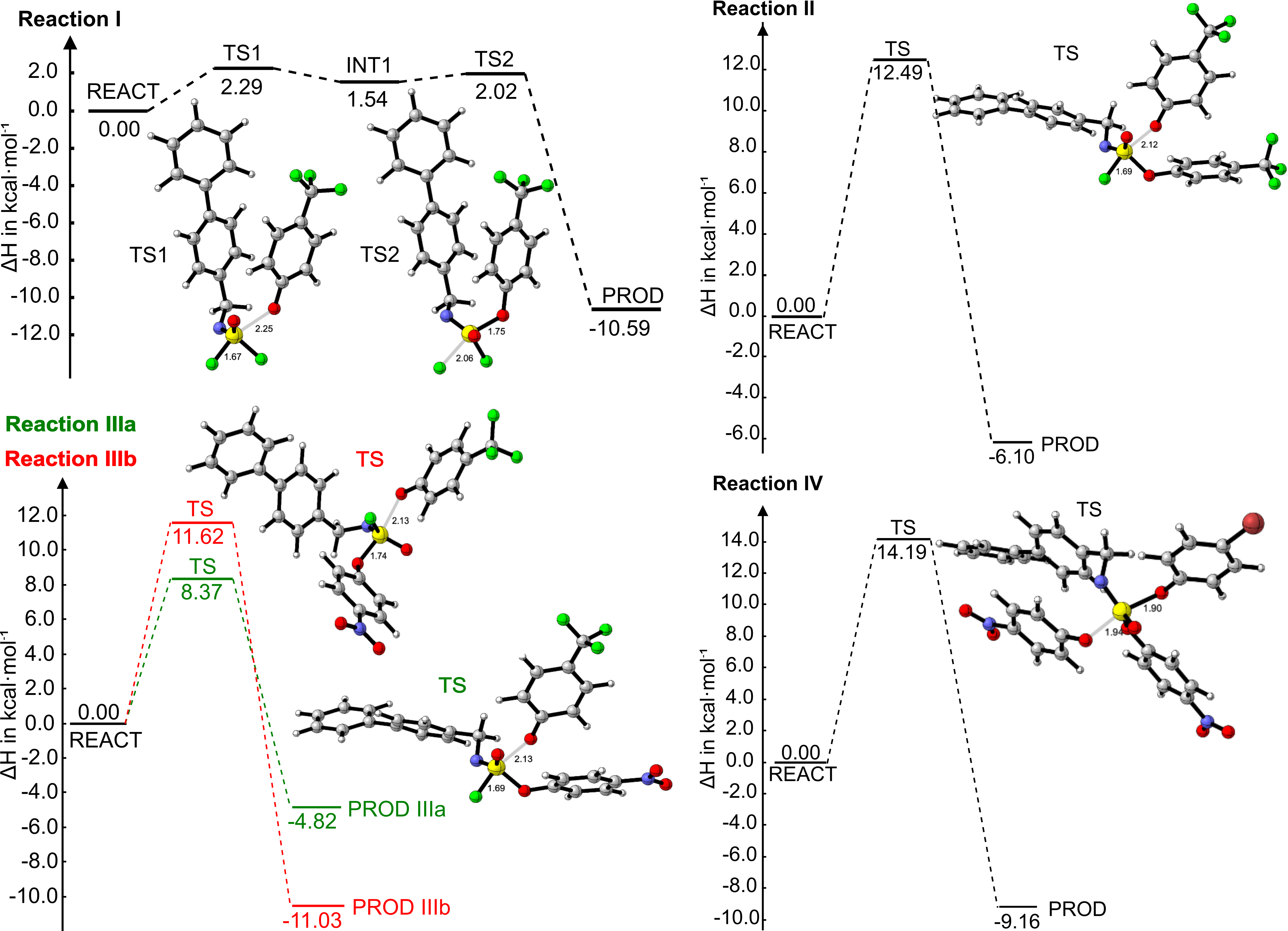
For years, the key to speeding up click reactions was thought to be lowering the activation enthalpy (ΔH‡). Our detailed kinetic and computational studies of SuFEx and SuPhenEx reactions revealed a surprising twist: activation entropy (ΔS‡), a measure of molecular disorder, often plays the dominant role in determining the reaction rate. In some cases, the reaction is controlled by a mixture of both factors, while in others, it is dominated by entropy alone. This finding challenges a long-standing belief and highlights the importance of considering entropic effects in the design of new and more efficient chemistries.
To better understand the forces at play, we needed to isolate the enthalpic contributions. Using Density Functional Theory (DFT) and the Activation Strain Model, we analyzed a series of SuPhenEx reactions. We discovered that in the gas phase, the reactivity is controlled by the HOMO-LUMO orbital interactions, which can be precisely tuned by changing the electronic substituents. In polar solvents, the control mechanism shifts, and electrostatic stabilization becomes the dominant factor. These insights provide a clear roadmap for experimentally designing more efficient S(VI) substitution reactions by controlling the enthalpic component.
Building on our detailed understanding of enthalpy, my future work is focused on the more elusive factor: entropy. By using a combination of Ab Initio Molecular Dynamics (AIMD) and Machine Learning (ML), the goal is to simulate and predict the entropic contributions to these reactions. By modeling the real-time, dynamic dance of molecules as they react, we can build predictive models that will allow for the rational design of reactions where both enthalpy and entropy are optimized for maximum efficiency and control.
… and more.